Understanding Surface Plasmon Resonance
Fundamentals of Surface Plasmon Resonance (SPR) Video (5 mins.)
Accelerating Biotherapeutic Discovery
Using High Throughput SPR to Explore the Full Kinetic and Epitope Diversity of Large Antibody Libraries
Making medicines is an expensive, time-consuming, and high-risk challenge, while also being a lucrative business. Though the cost of failure is significant in terms of both time and money, the reward can be incredibly valuable. Therefore, any method or technique that can minimize time and costs, while increasing the chance of finding and developing a successful therapeutic agent, is highly sought-after.
The search for biotherapeutics in the form of monoclonal antibodies(subsequently referred to as antibodies) is especially labor intensive. However, the naturally high affinities and exquisite target specificities found in this class of molecule provide unique therapeutic benefits to patients, making antibodies a worthwhile investment even when considering the various challenges that complicate their development. Many successful pharmaceuticals are small molecules, developed through a process of rational design and engineering to optimize desirable characteristics, including binding affinity and specificity.
In the case of antibodies, however, epitope specificity is an innate property that cannot be designed rationally, requiring empirical investigation. Discovering an antibody with appropriate characteristics relies upon an informed selection from the large antibody libraries produced using modern methods. Characterization of the binding interaction between the target antigen and therapeutic antibody candidates is an important step in this process, aiding in the selection of candidates to progress and helping to determine the crucial mechanism of action (MOA). High throughput SPR systems make possible the evaluation of large sets of antigen: antibody interactions quickly and cost-effectively by adding throughput to the proven surface plasmon resonance technique. This technology can be used to run parallel investigations into kinetics, affinities, and epitope specificities from the very start of the drug discovery process.
This ability to perform higher throughput and comprehensive characterization early in the drug discovery workflow changes the paradigm of therapeutic antibody screening. Researchers become better informed, earlier, to fully appreciate the epitope landscape of a campaign and so identify the superior candidates from the original library.
Before the advent of high throughput surface plasmon resonance screening, researchers commonly used high throughput but low information content assays to triage a subset of clones for subsequent detailed characterization. This approach meant underappreciating the full epitope and kinetic diversity of the original library. As a result, under-sampled pools led to picking suboptimal clones, which then required extensive engineering in the hope of improving their properties; or completely missing highly valuable potential therapeutics altogether. High throughput surface plasmon resonance screening avoids a great deal of redundant work, saves time and cost, and ultimately results in superior leads. Importantly, it also avoids researchers repeating or abandoning screening campaigns unnecessarily when they failed to identify desired clones; not because they weren’t in the library but because it was not possible to look deep enough, early enough. In this white paper, we look at the specific challenges surrounding the discovery of therapeutic antibodies and present high throughput solutions for antibody characterization.
High throughput surface plasmon resonance provides more information sooner, improving decision making and changing the paradigm of antibody screening. The result is faster and highly informed candidate selection/progression with a reduced risk of failure, increased intellectual property (IP) understanding, and minimized sample consumption. These properties make it particularly attractive to early-stage research where large numbers of samples are available for testing, but in limited quantities.
What makes a successful medicine?
One of the main factors necessary for a successful therapeutic agent is a well-defined MOA in the human body. This is an important consideration for safety and efficacy, and therefore for regulatory approval and clinical success. In addition, knowing and understanding a unique MOA is an essential part of securing IP rights and supporting successful commercialization.
A medicine can take many forms, from small molecules to vaccines and antibodies. In an ideal world, whether small or large molecule, all medicines should be straightforward to manufacture and produce consistent and predictable results, both in production and the clinic. For the patient, medicines should be safe, effective, and as convenient as possible to dose, in terms of size, frequency, route of administration, and cost.
Why antibodies?
The field of therapeutic antibodies is producing massive and growing sales for the pharmaceutical industry, with the number granted first approval in either the European Union or the United States reaching double figures for the first time in 20171. This accelerated pace is projected to continue through 2018 and beyond, cementing antibody therapeutics as an increasingly important part of the pharmaceuticals business, representing over 40% of all newly approved drugs.
Antibodies are highly attractive as therapeutic agents as they have a naturally high affinity and specificity for their target antigen. These are desirable characteristics that enable an antibody to bind well to a single target, maximizing the desired biological effect, while minimizing off-target issues. In turn, this means that antibodies can be dosed at relatively low levels compared with small molecules.
Furthermore, because of their large size, they are cleared from the body much more slowly than small molecules, so they can be administered less often (typically once a month), providing substantial patient convenience.
Adalimumab (marketed by AbbVie Pharmaceuticals as Humira) was the first fully human antibody granted US FDA approval and serves as an example of a highly successful therapeutic antibody. It is now used to treat several diseases and conditions including rheumatoid arthritis, Crohn’s disease, and ulcerative colitis. In 2017 AbbVie reported $18.427 billion USD of sales from Humira alone2.
The nature of antibodies
In mammals, an antibody is a specialized, Y-shaped glycoprotein produced and secreted by B lymphocytes in the immune system of an organism. Its primary function is to recognize and bind a unique pathogen (via an antigen) and neutralize it. The recognition and precise binding by the antibody to an antigen occurs via the antigen-binding sites located at the tips of the Y-shaped molecule’s Fab fragments. These regions provide a complex interface that recognizes and binds to a specific site on an antigen – termed an epitope.

“It is not likely that in silico tools/algorithms will ever be predictive enough to replace empirical selections in my career – the math is too daunting. No technology can exhaustively sample all the theoretical sequence space of an antibody molecule, let alone screen that diversity. Combine that with the huge diversity of possible antibody: antigen binding conformations and it seems like there will always be a need for actually generating variant libraries and screening them.” -Bill Harriman – VP, Antibody Discovery Services at Ligand Pharmaceuticals
The architecture of an antibody’s antigen-binding site is composed of stretches of conserved framework amino acid residues alternated with hypervariable portions, termed complementarity-determining regions (CDRs). As a result, an organism’s antibody repertoire is vast, with enormous sequence and structural diversity, enabling it to recognize an equally enormous range of antigen epitopes.
It is this virtually limitless target range and unique process of target recognition and binding that is of great interest when making medicines. The ability to develop an antibody capable of binding to a specific target of interest with its trademark high affinity and specificity is a model solution to drug discovery.
Discovering an antibody
Making a medicine, whether a small molecule or biologic such as an antibody, is not a prescriptive process where the end, target molecule is known. Rational design and empirical science represent two polarized, yet complementary approaches to drug discovery.
In the case of therapeutic antibodies, some characteristics, such as affinity, can be optimized via engineering, but it is not possible currently to predict or design the epitope specificity in a rational way. Rather, this is an innate property, which dictates binding to the target of interest (antigen). It cannot be engineered by design in silico and/or de novo synthesis.
Empirical science is therefore critical for the discovery of antibody therapeutics, and can be aided, but not replaced by computational methods.
A collection of antibodies can be functionally screened to determine which potential drug candidates to move through the pipeline. This method of using empirical evidence to ‘triage’ the antibodies available and select promising candidates is labor- and time intensive, as well as expensive. However, this experimental selection step is also crucial to the discovery process.
Some of the routine methods for triaging, such as ELISA, FACS, and kinetic studies, result in information being missed. However, the combination of modern antibody generation methods to create large diverse libraries and high throughput analytical tools for screening make it possible to carry out more comprehensive screens.
Library-to-leads triaging
Current techniques for generating antibodies include the use of hybridoma technology in normal and transgenic animals, phage display, and synthetic libraries. All these can produce prolific numbers of clones to feed drug discovery pipelines with increasingly large panels of potential therapeutic antibodies. A library produced by phage display, for example, typically includes over ten billion clones, while the current SuperHuman 2.0 library from Distributed Bio contains almost 100 billion unique members.
This colossal number of clones far exceeds the practical capacity of current expression and analytical methods, but rather, provides a diverse library from which binder clones towards a target of interest can be extracted, via panning strategies. This typically yields 100’s to 1000’s of hits, depending on the screening strategies used, which are routinely whittled down to a subset for detailed characterization.
Greater numbers of candidates increase the possibility of finding both an optimal antibody to treat a disease, and one which presents a good starting point in IP free space. This is crucial for selecting the most successful and commercially attractive medicine.
The process of deciding which antibodies to progress through the pipeline is termed library-to-leads triaging. Ideally, this triage step should take place in the early stage and examine the largest number of antibodies possible, generating information on as many binding interactions as practically possible to fully appreciate the epitope landscape of the campaign and minimize the need for further engineering.
High throughput analytical tools allow a broader and deeper diversity of the library to be discovered from the beginning of the process, enabling the identification of suitable candidates for progression, and reducing the need for extensive and costly engineering. Commercially, the generation of antibody libraries is a substantial investment. For example, a new line of transgenic animals can involve costs of greater than $1 million USD. Add to this the hugely competitive nature of drug discovery, where multiple large pharmaceutical and numerous biotechnology companies are often working on the same drug target, and the importance of rapidly realizing the full value of a purchased or generated antibody library to maximize output, return on investment, and identify/defend IP space is business critical.
“With small libraries, you struggle to identify weak hits with the characteristics you are interested in, and then spend a lot of time attempting to optimize them. Large libraries change this paradigm by ensuring that you have swarms of hits with any characteristic you are interested in, and don’t have to optimize them. Our new libraries have 1000x more molecules than old libraries and being able to find what you want up front is the core advantage.” Jacob Glanville – Founding Partner, Chief Science Officer, and Chairman of the Board at Distributed Bio
A closer look at the epitope
The library-to-leads triage process involves characterizing the binding interactions between candidate antibodies and target antigen. The physical contacts occurring within the antigen: antibody binding interaction form the epitope: paratope interface, where epitope refers to the contact residues contributed by the antigen and paratope refers to those contributed by the antibody. It is this interaction, and therefore the epitope itself, that defines an antibody’s MOA, making it important and relevant to the success of a therapeutic antibody program. A therapeutic antibody’s epitope specificity is a critical characteristic, and defining it is crucial in terms of IP rights in the fiercely competitive world of pharmaceuticals, where multiple companies are often working on the same targets to become first-in-class and/or best-in-class.
For example, although there are five antibodies targeting the PD-1 (programmed cell death-1) pathway already on the market to treat cancer, it remains a popular target for development. A further nine antibodies targeting PD-L1 (programmed death-ligand 1) are currently in clinical development1. Multiple companies also have antibodies targeting CGRP (calcitonin gene-related peptide) either on the market or in late-stage clinical trials for the prevention of migraines.
The ability to claim a novel epitope offers both a competitive edge and IP opportunities. Surveying the epitope landscape of a large antibody library at the earliest possibility, through functional library-to-leads triaging, is advantageous to this end, providing the opportunity to expand and defend IP space. Importantly this technology ensures that only those antibody clones exhibiting the most appropriate epitope specificity for commercial and clinical success are progressed.
Characterizing binding interactions: kinetics, affinity, and epitope specificity
The combination of three key characteristics – kinetics, affinity, and epitope specificity – provides a full understanding of antibody binding interactions. Each is important in isolation, offering specific and useful information for the antibody’s characterization. In combination, the three properties help to enable highly informed selection decisions.
Antibody Libraries IN FOCUS
Why does antibody discovery require the generation of large libraries?
Antibodies are incredibly diverse molecules. While small molecules often bind weakly and with limited specificity to an intended target, it is possible to generate antibodies with high affinity and specificity. These highly desirable properties are rare and therefore require much larger libraries as starting points for their discovery. In antibody discovery, there is always a need to generate many hits through sampling a large amount of sequence space and then sort through them quickly to generate optimal leads.
How large is large enough?
The bigger the library, the better. Antibody generation techniques have made it possible to create libraries with up to 100 billion unique members that feed the panning strategies against the target of interest to yield binder clones, typically 100’s or 1000’s of on-target hits.
What are the key benefits of using large libraries?
The availability of such large libraries has resulted in a change of paradigm – instead of finding 10-50 hits that require significant engineering, thousands of hits are possible, from which those with the desired features need to be selected.
Binding kinetics and affinity data
The strength of a binding interaction between two molecules, such as an antibody and its specific target, is characterized by the association and dissociation kinetic rate constants (ka and kd), which describe respectively the formation and decay of the bimolecular complex. The ratio of these values gives the equilibrium dissociation constant (KD= kd/ka). This describes the binding affinity of the molecules for one another and influences the recommended dose of a medicine.
Binding kinetics and affinities are important parameters in drug discovery for understanding an antibody’s MOA and can influence its pharmacodynamics and pharmacokinetics. Additionally, the binding kinetics provide useful information for guiding the evaluation of an antibody’s performance throughout the entire drug discovery and development pathway – from screening to manufacture.
Early-stage screening often does not include the generation of kinetic data because of the limited throughput of commonly used methods such as SPR, meaning that they are used as a secondary step after large antibody libraries have already been screened and triaged into smaller subsets. This is due to the large numbers of antibodies that require empirical investigation and the resultant need to reduce the processing burden wherever possible, often necessitating compromise on throughput or accuracy4.
A typical example of this approach might involve preliminary screening with a high throughput but low information (end-point) content assay, such as ELISA. Potentially important data is missed at this stage, increasing the likelihood that otherwise lucrative candidates are rejected through less than fully-informed decisions on pipeline progression.
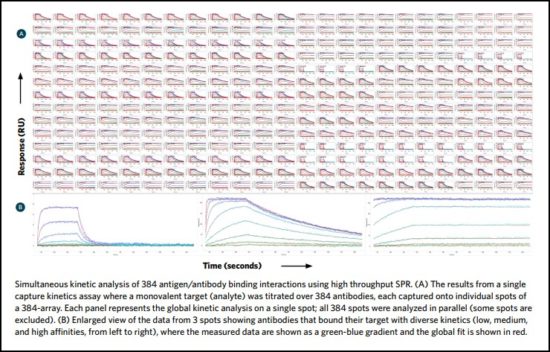
Epitope binning
Epitope binning – the empirical investigation of an antibody’s epitope specificity – involves clustering antibodies that bind to the same or similar epitopes into families, or “bins.” It is a competitive assay that tests antibodies in a pairwise and combinatorial manner for the ability to bind simultaneously to their specific antigen.
Should both antibodies bind at the same time, it is presumed that they are targeting distinct, non-overlapping epitopes. If one antibody blocks another, the inference is that they compete for overlapping or
similar epitopes.
In this way, all antibodies in the library are tested against each other, grouped according to their epitope specificity, and a “blocking footprint” generated for each. Antibodies sharing a bin are likely to also share similar binding interactions. Antibodies that exhibit unique epitopes can be rapidly and easily identified to provide diversification of selected candidates.
Performing epitope binning early in the discovery pathway using a large library of antibodies, rather than a subset, ensures that you see the full picture of all interactions. As a result, it is possible to identify binding pairs that would likely be missed if only a limited, smaller pool of antibodies were investigated.
Epitope binning can also reveal sandwiching antibody pairs that target different epitopes to the functional one and are therefore useful as companion reagents and/or diagnostics. This helps to support the therapeutic antibody discovery program, which involves numerous antibody-based assays throughout preclinical and clinical development. These further applications for antibodies – as diagnostic tools, companion diagnostics and reagents – can also provide large additional financial opportunities.
“Exploring the depth and breadth of an antibody library’s epitope coverage requires information on as many antibodies as possible to simultaneously probe all targeted epitopes and their relationships to one another. Performing these studies on large antibody arrays enables exquisite discrimination of near-identical clones and provides an efficient way of surveying the entire epitope landscape. Increasing the number of antibodies interrogated increases the resolution of the binning results, which in turn increases the chances of finding antibodies with unique or nuanced binding modes to strengthen IP.
High throughput SPR is an ideal tool for high throughput early-stage epitope characterization, revealing the full epitope repertoire of an antibody library, where clones are numerous but available only in
limited quantities.” Yasmina Noubia Abdiche – CSO, Carterra Inc.
Label-free biosensors
As discussed, the large number of antibodies present in a starting panel necessitates the use of an analytical tool to screen outputs and help triage library-to-lead candidates. Many available methods are slow, resource-intensive, and/or complicated. Researchers are therefore turning to label-free biosensors, such as SPR and BLI (biolayer interferometry) instruments. These are ideally suited to measure binding in real-time and without the need to conjugate or modify either partner.
SPR and BLI instruments can screen numerous clones from crude samples and are routinely used in the pharmaceutical industry to characterize antibody: antigen interactions. However, these techniques have limited throughput and require complex assays and relatively large sample volumes, meaning that they are often only used as a secondary screen.
High throughput mAb characterization using high throughput SPR for kinetics and epitope studies
Increasing the potential throughput of label-free biosensor technology would be a major development for those involved in antibody discovery. Additionally, performing both kinetic and epitope binning studies on a complete antibody library would allow the full interrogation of epitope diversity.
High throughput SPR systems, such as Carterra’ s LSA™ instrument, meet both demands. This technology rapidly and easily delivers throughput orders of magnitude higher than other label-free methods, making it possible to analyze many hundreds of interactions in parallel. This enables both kinetic screening and epitope binning studies of large antibody panels, ideally positioning them for early-stage screening. This ability to identify a few high-quality leads with relevant epitopes so early in the discovery process is invaluable to antibody discovery teams.
In terms of kinetic analyses, the capability to perform high throughput experiments with antibodies is important due to their uniquely tight binding with antigens. Compared to small molecules, which have generally transient binding interactions, antibody: antigen interactions often exhibit affinities that are a million-fold tighter. Accurately measuring kinetic rate constants of such high-affinity reactions, therefore, requires long binding cycles6. As a result, the ability to increase throughput is especially beneficial to achieve overall gains in efficiency and cost savings.
With Carterra’s LSA instrument, it is now possible to measure the binding kinetics of up to 1,152 antibody interactions in a single unattended run (3 x 384-well plates). High throughput surface plasmon resonance technology is thus gaining popularity for accelerating the kinetic screening of large antibody libraries, demonstrated by recently published studies.
Epitope binning experiments ideally involve the pairwise comparison of each antibody to every other antibody in the panel, as well as itself, and not just a preselected subset. Consequently, the number of interactions required scales geometrically with the number of antibodies in the test panel. For example, analyzing ten antibodies requires 100 interactions, while analyzing 100 antibodies requires 10,000 interactions.
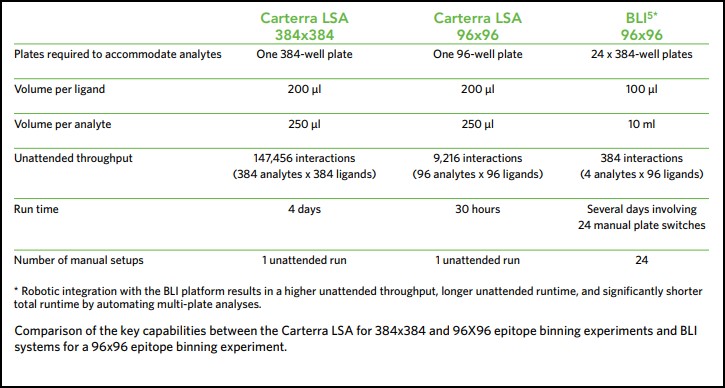
When exploring the epitope coverage of large antibody panels, the required number of interactions to perform a comprehensive binning analysis can escalate quickly and become prohibitively high, making throughput and sample consumption vital considerations for epitope binning investigations. This makes such experiments unfeasible on standard biosensor platforms. However, Carterra’s LSA instrument makes it possible to perform up to a 384×384 epitope binning experiment, equating to over 147,000 interactions, in a single run and using minimal sample volume.
The “one-on-many” array format employed by high throughput surface plasmon resonance means that the sample consumption per antibody does not scale with the size of the antibody panel. The same amount of antibody and ligand (coupled) or analyte (in solution) is used whether the array contains one or 384 antibodies, a significant practical advantage over other technologies.
Comparison to other label-free biosensors
It is the throughput capabilities and corresponding low sample consumption that set the Carterra LSA platform apart from other systems on the market. This is demonstrated in several recent studies comparing different label-free biosensor systems in terms of accuracy, throughput, and other key parameters.
The Biacore® 4000 from GE Healthcare (an 8-channel SPR instrument) and the MASS-1 system from Sierra Sensors (8-channel SPR instrument with hydrodynamic isolation (HI)) produce comparable kinetic rate constants for antigen/antibody interactions. However, these systems have limited throughput, being restricted to only eight, or sixteen interactions at a time respectively. Even the more recent versions of these systems, the Biacore 8K and MASS-2, can only provide eight and thirty-two interactions at a time respectively. The Carterra LSA platform generates equivalent kinetic rate constants and data quality, but facilitates the rapid generation of kinetic screening data from up to 384 samples in parallel and 1,152 samples per unattended run, representing the highest throughput available on the market.
Another recent study compared the Octet-HTX from Forte Bio/Pall to a high throughput surface plasmon resonance platform for epitope binning of 96-antibody arrays. The Octet-HTX uses BLI technology, which is highly flexible and provides the ability to simultaneously analyze interactions of 96 independent analyte/ligand pairs. However, since sample consumption scales with the size of the antibody panel, it is often only possible to perform investigations with relatively small panels due to the high demand for material, complicated layout, and the additional robotic integration needed to automate multi-plate feeding. This limits the potential for large-scale early stage antibody screening with BLI technology. By comparison, high throughput surface plasmon resonance provides a method that enables the simultaneous analysis of 384 binding interactions and requires an exceptionally low sample consumption, making high throughput epitope binning assays possible11. The Carterra LSA can perform a comprehensive 384×384 binning assay in a single unattended run.
Despite challenges in their discovery, antibody therapeutics have proven to be highly effective and commercially valuable medicines. Standard characterization technologies require compromises in workflow that ultimately reduce the effectiveness of antibody discovery. Streamlining the library-to-lead process through advanced technology helps deliver medicines faster, cutting costs and increasing commercial value.
The advent of high throughput surface plasmon resonance instruments, such as the Carterra LSA platform, now allow researchers to realize the full kinetic and epitope potential of their large antibody libraries earlier in the drug discovery process. Positioned upstream relative to traditional lower throughput surface plasmon resonance platforms, high throughput surface plasmon resonance combines both high-throughput and high-resolution analysis to enhance screening methods and permit the rapid identification of unique, highly sought-after antibody clones for clinical and commercial benefit.
We would like to thank Bill Harriman (VP, Antibody Discovery Services at Ligand Pharmaceuticals) and Jacob Glanville
(Founding Partner, Chief Science Officer, and Chairman of the Board at Distributed Bio) for their help with developing this
white paper.
Discovering an Antibody’s Therapeutic Fingerprint
Utilizing Multi-parameter Epitope Binning to Understand a Therapeutic Antibody’s Mechanism of Action
Antibodies represent the fastest growing class of drugs in the market today, with over 60 antibody drugs currently approved for use and more than 550 in clinical development. In 2017, the number of antibodies granted a first approval in either the European Union (EU) or United States (US) reached double digits for the first time, a trend that continued into 20182.
The discovery and development of therapeutic antibodies therefore represents an especially lucrative opportunity for biopharmaceutical companies worldwide, as demonstrated by Adalimumab marketed by AbbVie Pharmaceuticals as Humira), the first fully human antibody to be granted approval by the US Food and Drug Administration (FDA). It serves as a benchmark for the commercial potential of therapeutic antibodies, with considerable success in the treatment of several autoimmune diseases and conditions including rheumatoid arthritis, Crohn’s disease, and ulcerative colitis. Humira’s reported sales of almost $18.5 billion USD annually in 20173 establishes it as the top blockbuster drug in terms of sales.
While it can be incredibly valuable both in terms of advancing human health and increasing profits, the search for the next blockbuster therapeutic antibody is expensive, time-consuming, and has a high failure rate. New technologies that help minimize risk to pharmaceutical companies and increase the chance of finding “the next Humira” are highly sought-after.
High throughput Surface Plasmon Resonance (SPR) technology has recently facilitated a paradigm shift in the discovery of therapeutic antibodies, enabling the rapid and detailed characterization of large antibody libraries at the earliest stage of the process—so-called high-information-content screening. This allows researchers to expand their screening efforts to much larger libraries, learn about antibody: antigen binding interactions in greater depth, and combine orthogonal data to create a highly informative fingerprint of the antibodies of interest. Composing a detailed picture from multiple sources of information enables informed lead selection and helps to determine an antibody’s mechanism of action (MOA).
With many of the best understood antibody drug targets already extensively exploited in drug development, identifying antibodies with novel MOAs is the key to discovering and marketing new biotherapeutics and extending applications. This is critical in the competitive world of pharmaceuticals and intellectual property (IP) rights.
In this white paper, we first look at antibody libraries and the importance of understanding their binding properties in detail. High throughput SPR provides a method for characterizing the binding kinetics, affinities and epitope specificities of large antibody panels, and when these data are merged with the results from other assays, they can aid in determining MOA. We will outline the process of antibody fingerprinting and describe a case study where combining epitope binning with orthogonal data enabled the identification of human antibodies responsible for neutralizing the pathogenic activity of Staphylococcus aureus (S. aureus) via two distinct MOAs.
Understanding your antibody library
The importance of the epitope
The primary evolutionary purpose of an antibody is to recognize and bind its antigen—historically an invading pathogen—with appropriate affinity and specificity to neutralize it. A specific site on an antigen, termed an epitope, is recognized and bound by so-called antigen binding sites located at the tips of the Y-shaped antibody molecules, termed the paratope. The complex and hypervariable architecture of these antigen-binding sites enables a vast range of epitopes to be recognized by an organism’s antibody repertoire. It is this virtually limitless target range, combined with the trademark high affinity and exquisite target specificity of antibodies that is of great interest for biotherapeutics.
The ability to develop an antibody de novo that can bind to a specific target of interest would be a model solution to drug discovery. However, while all other characteristics of an antibody drug can be optimized by engineering – such as the binding kinetics, affinity, and thermostability – it is currently impossible to apply rational design and engineering to epitope recognition and binding, as epitope specificity is an innate property that cannot be predicted. The epitope is highly complex and unknown a priori, and the in silico design of an equally complex complementary paratope on the antibody presents a significant challenge to computational biologists. The discovery of an antibody with appropriate characteristics therefore relies on informed, empirical selection from large antibody libraries.
Epitope specificity is also critical for dictating an antibody’s biological function and MOA, providing the means to recognize, interact, and affect a biological mechanism. The discovery of antibodies with unique epitope binding characteristics and MOA is therefore the critical step in successful biotherapeutics research to differentiate an IP portfolio from the competition.
Antibody screening: Then and now
A therapeutic antibody discovery program involves functionally screening large antibody libraries that could potentially contain billions of unique clones. Through an empirical triage process, promising candidates are selected and progressed through the development pipeline. However, as a result of modern antibody generation techniques, extremely large and diverse libraries are created. While this is beneficial for increasing the possibility of finding a desirable and optimal antibody in the original library, it also creates major practical challenges in terms of their screening. Before the advent of high throughput SPR, drug discovery scientists would commonly use high throughput but low-information-content assays (e.g. ELISA and FACS); or low throughput but high-information content SPR to triage only a subset of clones from a library. Specifically, these techniques do not provide any information on the epitope coverage of the library, with detailed characterization only used as a secondary step. This approach underappreciates the full epitope and kinetic diversity of the original library, resulting in undersampled pools and the possible selection of suboptimal clones that require extensive engineering. It is also likely to miss potentially high-value antibody drugs completely, or eliminate them from the sample pool prematurely.
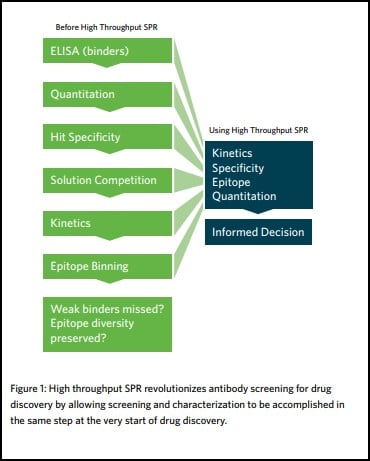
High throughput surface plasmon resonance revolutionizes antibody screening for drug discovery by allowing screening and characterization to be accomplished in the same step. By expanding the throughput of SPR, a de facto technique for delivering exceptional resolution and high data quality, it can now be positioned upstream in the drug discovery process, which means it can be used to triage a much larger antibody panel.
Researchers become better informed, earlier, and can carry out detailed studies of the binding interactions between the target antigen and large antibody libraries. Information on antibody: antigen binding interactions, such as their kinetics, affinity, and epitope specificity, provide researchers with comprehensive information that guides decisions on which antibodies to follow up on with other assays.
Why do we need to know a specific antibody’s MOA?
In pharmacology, MOA describes the specific biochemical interactions that occur for a drug to produce an effect. MOA is driven by the molecular binding properties of a candidate drug and how it interacts with its specific (intended) and unspecific (unwanted) binding partners in terms of its kinetics, affinity, selectivity and epitope specificity.
Elucidating the MOA of any novel drug, including therapeutic antibodies, is important for several reasons. Knowledge of the MOA drives better-informed decisions that impact both the safety and efficacy of a drug. Greater understanding of MOA helps in predicting a patient’s likeliness to respond to a particular treatment. The knowledge gained can also be used to decide how best to combine drugs for the treatment of diseases such as cancer, in order to reduce risks of resistance and treatment failure. In addition, understanding MOA is advantageous in the development of new drugs, either highlighting an important molecular interaction and/or target for further drug development; or identifying novel MOAs that offer IP and commercial potential.
With high throughput SPR, it is possible to survey the epitope landscape of an antibody screening program during early-stage research, where large numbers of clones are available in low quantities. This provides a functionally-relevant way of navigating an antibody panel to guide the triaging of hits and prioritize resources to determine MOA.
Multi-parameter epitope binning
Epitope binning using high throughput SPR
Epitope binning using high throughput surface plasmon resonance is an incredibly powerful tool in guiding the discovery of therapeutic antibodies because it can be used to organize antibodies into families based upon their epitope specificities. Using high throughput surface plasmon resonance, epitope binning experiments can be performed at the earliest stage of drug discovery to assess an antibody libraries epitope coverage.
High throughput surface plasmon resonance is the only technique that can deliver the throughput and multiplex capabilities that provide comprehensive data to enable epitope binning studies and resultant MOA fingerprints. The Carterra® LSA™ high throughput SPR platform can perform these experiments in unprecedented high throughput, with the added benefit of minimal sample consumption, making these assays amenable to early-stage research and discovery. Furthermore, Carterra’s proprietary Epitope Data Analysis Software enables the results from several orthogonal studies, such as structural or mutagenesis data, to be merged with the epitope binning results, producing a detailed fingerprint (Fig. 3). This provides a highly informative visualization tool that aids researchers in decision-making throughout the drug development process.
Using the Carterra LSA for multi-parameter epitope binning
The unique, high throughput capabilities of the Carterra LSA are realized through proprietary technology that enables a panel of up to 384 antibodies to be arrayed and reloaded onto a single chip. Antibodies are immobilized as ligands to produce a 384-ligand array per chip.
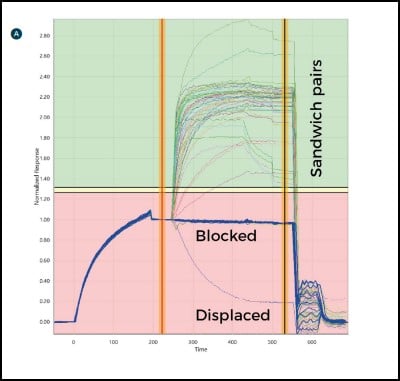
Antibodies are then injected in series as analytes to test for sandwich pairing. Using this one-on-many analyte-on-ligand assay format, the LSA delivers real-time data on up to 384 ligands in parallel. In this format, all antibody analytes are tested for sandwich pairing with all antibody ligands, providing a comprehensive 384×384 interaction matrix and a detailed picture of the antibody panel’s epitope landscape.
Each time an antibody is injected as an analyte, the antibody spot is measured for any change in mass. Sensorgrams are produced that show the binding interaction of the antibody of interest with the antigen, and then with every other antibody in the panel in turn (Fig. 3). Since each binding cycle includes the entire 384-ligand array, in 384 binding cycles it is possible to rapidly perform a full 384×384 interaction analysis matrix and gain a comprehensive data set for all antibodies in the test set. Using the LSA, a typical 384×384 experiment can be completed in an unattended run within three days using very little sample (typically only 10 μg per antibody).
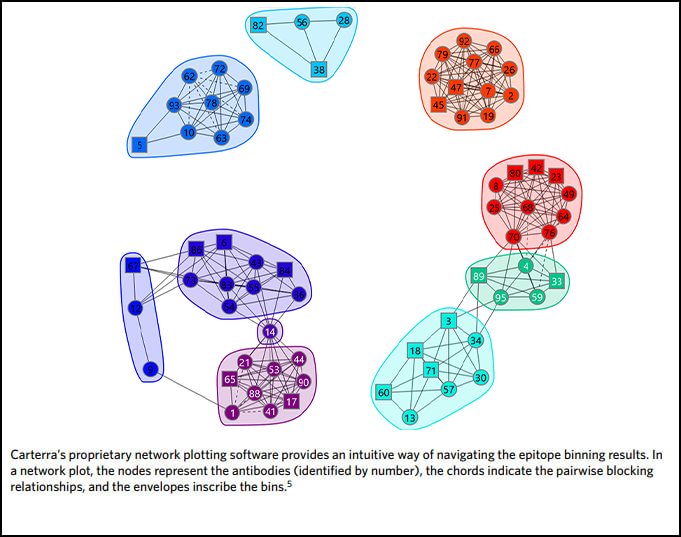
Carterra’s proprietary Epitope Data Analysis Software collates the data for all of the antibodies in an epitope binning experiment and combines this with data from orthogonal experiments (e.g. cell-based neutralization data, kinetics, affinity, mutagenesis) in a network plot (Fig. 3). In this way, an antibody fingerprint is constructed to help researchers improve efficiency and productivity for optimum outcomes.
Incorporating binding kinetics and affinity data
As well as epitope binning experiments, the Carterra LSA can perform high throughput and detailed kinetic and affinity determinations of up to 384 antibodies in parallel. Using the full capacity of the instrument’s autosampler it is also possible to load three 384 microtiter plates and determine full kinetic and affinity on up to 1152 unique clones in a single unattended run. These are important parameters in drug discovery for understanding an antibody’s MOA, pharmacodynamics, and pharmacokinetics; while the ability to screen large numbers has become crucial in the evaluation of the expansive libraries now
available.
The strength of a binding interaction between an antibody and its specific antigen is characterized by the association and dissociation kinetic rate constants (ka and kd). These constants describe the formation and decay of the bimolecular complex respectively, and the ratio of these values gives the equilibrium dissociation constant (KD = kd /ka). This describes the binding affinity of the molecules for
one another and influences the recommended dose of a medicine.
Early-stage screening often does not include the generation of kinetic data because of the limited throughput of traditional surface plasmon resonance or other label-free platforms. However, with the Carterra LSA, it is possible to perform kinetic studies at the first stage of drug discovery, combining affinity and binding data with results from epitope binning. As a result, researchers can start to determine an antibody fingerprint and build up a more complete picture of the antibody library as a whole, from the very start of the drug discovery process.
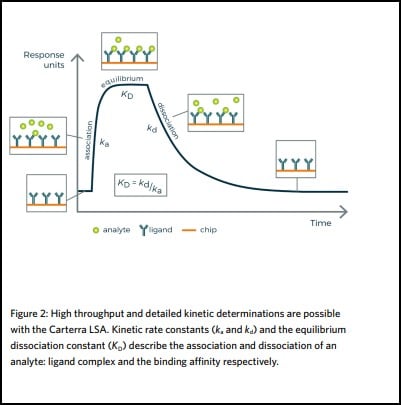
High sensitivity, high throughput surface plasmon resonance kinetics
Using the Carterra LSA instrument, it is possible to perform high throughput antibody screening and characterization in the same step and generate high quality, reliable kinetic data at the earliest stage of drug discovery.
The binding kinetics of up to 1152 antibody interactions can be determined in a single run due to Carterra’s patented flow printing technology, which also makes it possible to obtain data from low titer crude antibody preparations. These types of samples are common in early stage screening where in vitro antibody libraries are often supplied as crude periplasmic extracts (PPEs).
Despite the low expression levels of recombinant antibody fragments within these extracts (e.g. single-chain variable fragments (scFv)), the highly sensitive Carterra LSA can be used to measure the binding kinetics in high throughput. The LSA’s bi-directional or multi-pass flow for sample delivery extends the contact time during immobilization, generating multiple passes between sample and chip. This effectively concentrates material onto the chip surface, enriching capture levels of crude antibodies and therefore greatly enhancing assay sensitivity.
The Carterra LSA delivers kinetic data of comparable quality to other called gold-standard surface plasmon resonance platform, but with the added benefits of significantly higher throughput and the ability to detect lower PPE concentrations, while using less sample.
Combining orthogonal data to produce an antibody fingerprint
The key to multi-parameter epitope binning is the incorporation of data from additional orthogonal studies to identify functionality within bins, which aids in the development of the MOA fingerprint and the selection of clones with high therapeutic potential. The type of orthogonal data needed to determine functional activity depends somewhat on the system being investigated, however, mutagenesis, functional cell-based assays, cross-reactivity studies, and structural studies are all highly informative. While the specifics of a functional test are target dependent, epitope binning is agnostic of the type of therapeutic target or indication and is a universal test that can be performed on any antibody panel to understand its epitope coverage.
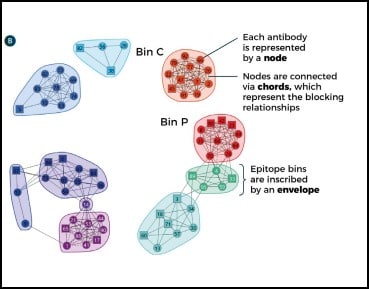
In some cases, orthogonal studies can be incredibly expensive and time-consuming. This is especially true for structural studies, such as the generation of crystal structures of antibody complexed with target antigen. These experiments are critical in confirming MOA but can be very complex, involving significant resources and large amounts of researchers’ time. By combining functional and epitope binning studies, it is possible to navigate the decision making of drug discovery scientists to those epitope bins of most interest prior to this step, thereby both speeding up the process and reducing costs. Carterra’s proprietary Epitope Tool data analysis software can be used to collate all data from a multi-parameter epitope binning experiment in a network plot, highlighting the bins of interest in terms of epitope and MOA. It contains a node for each antibody in the investigation, identifiable by its number. Nodes are connected to one another via chords to denote the blocking relationships. An envelope then inscribes antibodies that belong to the same bin–defined by sharing the same pattern of reactivity (blocking or sandwiching) against all other antibodies tested (Fig. 3). Once the bins are assigned, orthogonal data is applied by coloring the plot in different ways. In this way, a comprehensive fingerprint is produced that helps to identify those bins with similar functional effects for progression through to MOA elucidation studies.
Case Study: Neutralization of a Staphylococcus aureus virulence factor by the human antibody repertoire
Yeung et al. clearly demonstrate the power of multi-parameter epitope binning using High throughput Surface Plasmon Resonance for elucidating MOA in a recent paper published in Nature Communications4. In this section, we show how the process described above was applied to not only identify antibodies that neutralized a Staphylococcus aureus virulence factor but to differentiate two distinct mechanisms for neutralization.
Understanding the pathology
S. aureus is a major human pathogen that is also a commensal, with most individuals not experiencing a serious infection. However, it can cause significant morbidity and mortality through conditions such as bacteremia, pneumonia, and infective endocarditis among others5. The paper investigates the mechanism that S. aureus uses to steal iron from hemoglobin in a process necessary for its colonization and pathogenesis.
A protein called IsdB (iron-regulated surface determinant B) is central to this process. It is a surface-exposed protein anchored to the cell wall that removes heme from hemoglobin and transfers it to other Isd proteins. These, in turn, import and degrade it to release the necessary iron into the bacterial cytoplasm.
Anti-IsdB antibodies can confer protection against S. aureus infections in animal models, which makes this system and mechanism interesting to study not only to better understand the human immune repertoire since S. aureus is a commensal pathogen, but also in selecting therapeutic antibodies.
Molecular characterization of the antibodies of interest – combining epitope binning and orthogonal data
The researchers investigated recombinant anti-IsdB antibodies sourced by B-cell cloning from four healthy donors. The antibody repertoire was screened through epitope binning and studied in detail using a number of molecular characterization techniques. This enabled the antibodies to be organized into epitope clusters onto which the additional data was layered to create an antibody fingerprint that helps better understand the relevance of the epitope landscape.
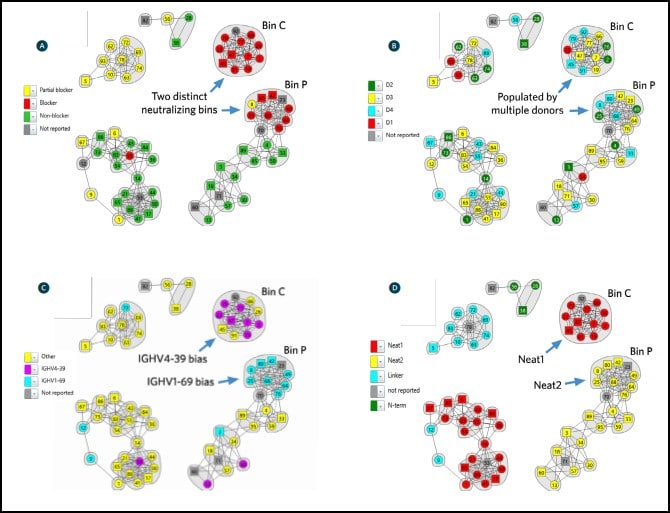
Epitope binning using high throughput surface plasmon resonance organized the antibodies quickly into epitope clusters. This study investigated 70 unique antibodies, which were screened in a pairwise and combinatorial manner yielding the network plot shown in Figure 4.
Once sorted into bin clusters, the researchers performed cell-based functional studies to determine whether the antibodies blocked IsdB’s interaction with its natural binding partner, hemoglobin, to neutralize S. aureus. Figure 4A shows that hemoglobin blockade was restricted to two distinct bin clusters, namely bin C and bin P (red nodes). Biosensor-based hemoglobin blocking assays also corroborated this result.
This observation suggested that two distinct MOA’s may be responsible for neutralization, which motivated further study.
Examining the source of the antibodies contained in bin C and bin P showed that they were populated by antibodies from multiple donors (Fig. 4B), which suggests that nature has found universal solutions to neutralization that are available in the human antibody repertoire. Remarkably, examining the genetic sequences of the antibodies contained in bin C and bin P revealed a strongly biased use of two immunoglobulin heavy chain germlines that neutralized IsdB (Fig. 4C), namely IGHV4-39 (bin C) and IGHV1-69 (bin P).
Antigen mutagenesis studies showed that bin C and bin P specifically targeted different IsdB NEAT (NEAr iron Transporter) domains, which are responsible for the binding of hemoglobin and heme respectively. Antibodies in bin C were shown to target NEAT1 and antibodies in bin P were shown to target NEAT2 (Fig. 4D). These observations are consistent with bin C and bin P providing neutralization via different MOA’s.
These results were confirmed by crystallography data, which showed at atomic resolution that antibodies in bin C, targeting NEAT1 primarily using IGHV4-39, used a near-identical binding motif to do so. Similarly, antibodies in bin P, targeting NEAT2 primarily using IGHV1-69, shared a near-identical binding mode.

Structural Studies for MOA While atomic-level structural data determined via crystallographic studies is the gold standard for defining an epitope, it is low-throughput, slow and resource-intensive. It is therefore used to confirm the MOA of high-value candidates rather than to predict or screen for them. In contrast, the results from high throughput surface plasmon resonance-based epitope binning studies enable a quick surveyance of an antibody panel’s epitope landscape, effectively clustering antibodies into epitope families. When merged with orthogonal data, such as functional cell-based neutralization data, antibody- sequence data, antibody source, and antigen mutagenesis studies (as shown in Fig. 4), the relevance of these epitope families is revealed, guiding the selection of high-value clones meriting further study.
Indeed, the study by Yeung et al. elegantly shows how multiparameter epitope binning enabled the identification of function blocking clones with two distinct MOA’s, which were then confirmed with structural studies.
The fact that the same phenotype of cell-based S. aureus neutralization could be achieved through two different MOAs is an important consideration in drug discovery, where pharmaceutical companies seek mechanistically-differentiated ways to solve a biological problem. This is important to both gain IP advantages over their competition and offer a safer or more efficacious medicine to patients.

With high throughput surface plasmon resonance, drug discovery scientists have the potential to perform powerful epitope binning experiments and detailed characterization of antibodies of interest at the earliest stage of the discovery process. This technology facilitates a paradigm shift in antibody discovery, identifying blocking relationships for a whole antibody panel from the beginning for a streamlined and highly-informed lead selection process.
The results from epitope binning provide the first layer of information in constructing a detailed fingerprint of an antibody library, where groups of antibodies are clustered together based upon their epitope specificity, which correlates to their functional activity. In other words, antibodies targeting the same or similar epitope are likely to share the same or similar MOA. Combining orthogonal data from functional and other studies builds on this layer-by-layer, helping researchers to navigate through the epitope landscape, prioritizing resources and characterizing binding interactions to aid in elucidating MOA. Using the Carterra LSA platform for high throughput surface plasmon resonance, it is possible to perform multi-parameter epitope binning to understand a therapeutic antibody’s mechanism of action—a critical feature in drug discovery and development campaigns.
References
1. Carter, PJ. and Lazar, GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nature Reviews Drug Discovery 17, 197-223 (2018)
2. Kaplon, H. and Reichert, JM. Antibodies to watch in 2018. MAbs 10(2), 183-203 (2018)
3. AbbVie Inc. AbbVie Reports Full-Year and Fourth-Quarter 2017 Financial Results. 2018 [Press Release] Available from: https://news.abbvie.com/
news/abbvie-reports-full-year-and-fourth-quarter-2017-financial-results.htm
4. Yeung, YA et al. Germline-encoded neutralization of a Staphylococcus aureus virulence factor by the human antibody repetoire. Nature
Communications 7:13376 (2016)
5. Tong, SY et al. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations and management. Clin. Microbiol. Rev. 28,
603-661 (2015)
6. Cassat, JE et al, Iron in infection and immunity. Cell Host Microbe 13, 509-519 (2013)
7. Abdiche, YN et al, Antibodies Targeting Closely Adjacent or Minimally Overlapping Epitopes Can Displace One Another. PLOS ONE (2017)
8. Kaplon, H. and Reichert, JM. Antibodies to watch in 2018. MAbs 2018 Feb/Mar; 10(2): 183-203
9. AbbVie Inc. AbbVie Reports Full-Year and Fourth-Quarter 2017 Financial Results. 2018 [Press Release] Available from: https://news.
abbvie.com/news/abbvie-reports-full-year-and-fourth-quarter-2017-financial-results.htm
10. Pellesi, L., et al. Spotlight on anti-CGRP monoclonal antibodies in migraine: the clinical evidence to date. Clin Pharmacol Drug Dev 2017
Nov; 6(6); 534-547
11. Estep, P., et al. High throughput solution-based measurement of antibody-antigen affinity and epitope binning. mAbs 2013 Mar/Apr; 5:2;
270-278
12. Abdiche, Y.N., et al High-throughput epitope binning assays on label-free array-based biosensors can yield exquisite epitope discrimination that facilitates the selection of monoclonal antibodies with functional activity. PLOS ONE 2014 Mar; 9(3); e92451
13. Katsamba, P.S., et al. Kinetic analysis of a high-affinity antibody/antigen interaction performed by multiple Biacore users. Analytical
Biochem 2006 May; 352(2):208-221
14. Adler, A.S., et al. A natively paried antibody library yields drug leads with higher sensitivity and specificity than a randomly paired antibody library. mAbs 2018 Jan; 29: 1-13
15. Ching, K., et al. Chickens with humanized immunoglobulin genes generate antibodies with high affinity and broad epitope coverage to
conserved targets. mAbs 2018 Jan; 10(1): 71-80
16. Kamat, V. and Rafique, A., Exploring sensitivity and throughput off a parallel flow SPRi biosensor for characterization of antibody-antigen
interactions. Analytical Biochem 2017 Feb; 525: 8-22
17. Sapparapu, G., et al. Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 2016 Nov;
540(7633): 443-447
18. Sivasubramanian, A., et al. Broad epitope coverage of a human in vitro antibody library. mAbs 2016; 9(1); 29-42